Non-Advanced Systemic Mastocytosis: KIT-Driven Mechanisms, Symptom Burden & Targeted KIT Inhibition
This page focuses on mechanism—how mutant KIT drives mast cell accumulation and mediator release—and how modern care is evolving beyond “stability”.
Key takeaways
- Non-advanced systemic mastocytosis (e.g., bone marrow mastocytosis, indolent SM, smouldering SM) can still carry a high day-to-day symptom burden despite absence of organ damage.
- For most patients, KIT D816V is the central disease driver, producing a constitutively active KIT receptor that supports mast cell proliferation and survival.
- Symptoms reflect both mast cell burden (tissue infiltration) and mediator release (histamine, tryptase and other inflammatory signals) affecting multiple organ systems.
- Targeted KIT inhibition (notably avapritinib (an oral kinase inhibitor) in indolent SM with moderate–severe symptoms) has randomised trial evidence showing improvements in patient-reported symptoms and objective markers such as serum tryptase. (See Targeted care.)
- “Stability” in modern SM care increasingly means measurable symptom control, improved quality of life, reduced mediator activity, and (in selected cases) reduced clonal mast cell burden, rather than simply “no organ damage”.
What is systemic mastocytosis?
Definition
Systemic mastocytosis (SM) is a rare clonal mast cell disorder characterised by the accumulation of abnormal mast cells in extracutaneous organs (commonly bone marrow, gastrointestinal tract, liver, spleen, and sometimes skin).
In cutaneous mastocytosis (often paediatric, skin-limited) with systemic disease (primarily adult).
Non-advanced vs advanced disease
- Non-advanced SM: typically bone marrow mastocytosis (BMM), indolent SM (ISM) and smouldering SM (SSM).
- Advanced SM: includes aggressive SM, SM with an associated haematologic neoplasm (SM-AHN), and mast cell leukaemia—defined by organ damage and poorer survival.
This webpage focuses on non-advanced SM.
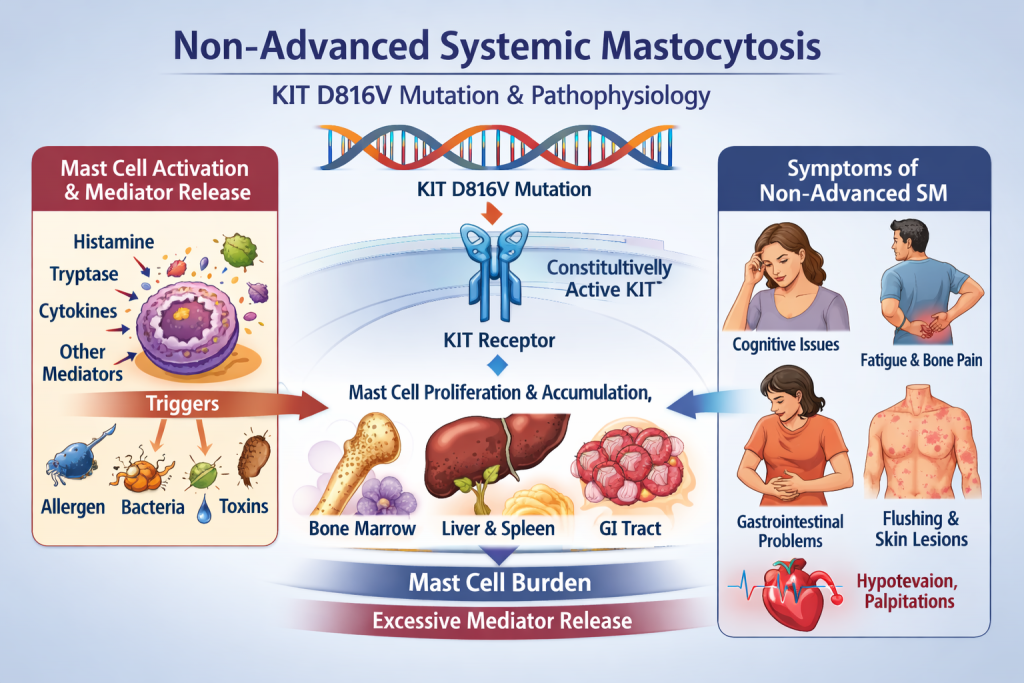
Mechanism: KIT D816V → mast cell burden → mediator biology
In most SM, the KIT D816V driver mutation creates an “always-on” KIT receptor that promotes mast cell expansion and survival; increased mast cells then release mediators (e.g., histamine/tryptase) that drive multi-system symptoms.
1) KIT as the disease “ignition switch”
KIT (CD117) is a receptor tyrosine kinase important in mast cell development. The KIT D816V mutation—present in the great majority of SM cases—
stabilises an active kinase conformation, producing constitutive signalling (even without ligand).
Practical implication: the mutation is both diagnostically important and therapeutically targetable.
2) Downstream signalling pathways
Constitutively active KIT can activate multiple pro-survival and proliferation pathways (often described in SM reviews as
MAPK/ERK, PI3K/AKT and STAT signalling), supporting accumulation of clonal mast cells across tissues. :contentReference[oaicite:0]{index=0}
- Burden effects: tissue infiltration (e.g., marrow, GI tract, liver/spleen).
- Mediator effects: dysregulated activation and release of bioactive mediators.
3) Mediators and why symptoms are “multi-system”
Mast cells can be triggered by allergens, infections, medications, physical stimuli and other inflammatory signals; mediators such as
histamine and tryptase (and many others) can drive symptoms across:
| System | Common symptom patterns described in SM | How mediator biology contributes |
|---|---|---|
| Skin | Flushing, pruritus, urticaria, red-brown macules/papules (if skin involved) | Histamine and other mediators increase vasodilation, itch and wheal/flare responses |
| Gastrointestinal | Cramping, diarrhoea, reflux, nausea/vomiting | Mediators alter secretion, motility and visceral sensitivity |
| Cardiovascular | Hypotension, tachycardia, presyncope; anaphylaxis risk | Vascular permeability/vasodilation and systemic mediator surges |
| Neurological / cognitive | Migraines, “brain fog”, anxiety/depression, impaired concentration | Neuroimmune signalling and systemic inflammation can worsen symptoms and sleep/fatigue |
| Musculoskeletal / bone | Bone pain; osteopenia/osteoporosis (and less commonly other bone phenotypes) | Inflammatory signalling plus marrow involvement can contribute to pain and remodelling |
Patients highlight fluctuating respiratory symptoms early on, later GI symptoms, near-constant headaches,
severe nightly bone pain, fatigue and cognitive issues—typical of a multi-system mediator phenotype.
“Beyond stability”: symptom burden and quality of life
The patients may be labelled “stable” because they have non-advanced disease and no overt organ damage, yet still experience daily, function-limiting symptoms.
What patients report in real life
- Unpredictable gastrointestinal urgency and dietary restriction attempts
- Chronic headaches/migraines and “brain fog”
- Severe bone pain and heavy reliance on analgesics
- Fatigue, reduced participation in family/social life
What “disease control” can mean today
- Symptom control measured with validated patient-reported tools (e.g., symptom severity scores)
- Reduced mediator activity (e.g., lower serum tryptase in responsive patients)
- Improved quality of life and functional participation
- Reduced mast cell burden (where achievable and clinically appropriate)
Epidemiology: Europe alongside the United States
Europe
- Orphanet cites European prevalence estimates for systemic mastocytosis around 1 in 7,700 to 1 in 10,400
- A Danish population study reported adult mastocytosis prevalence of 27.43 per 100,000 (as of 1 Jan 2022) and a 25-year average incidence of 1.21 per 100,000, with increasing incidence over time.
- A French estimate reported mastocytosis prevalence around 8.5 per 100,000.
Differences across European studies reflect case-finding, diagnostic access, coding practices, and whether cutaneous and systemic forms are combined.
United States
A commonly quoted US prevalence range for systemic mastocytosis on the order of ~1 in 5,000 to 1 in 10,000.
Real-world estimates vary by methodology, and under-diagnosis remains a concern, particularly where access to high-sensitivity KIT testing and tryptase measurement is limited.
Practical point: in regions without serum tryptase testing or sensitive molecular diagnostics, prevalence may appear lower simply because fewer patients are identified.
Diagnosis & classification (focus on non-advanced SM)
Diagnostic framework
SM diagnosis is based on meeting defined criteria using histology, immunophenotyping, molecular testing and serum tryptase.
Commonly used frameworks include WHO classifications, with a combination of major/minor criteria (e.g., dense mast cell aggregates on biopsy, KIT mutation, aberrant CD markers, atypical morphology, elevated tryptase).
Why tryptase interpretation may need nuance
Basal serum tryptase can be elevated for reasons other than SM, including hereditary alpha-tryptasaemia (HαT), a relatively common genetic trait associated with increased TPSAB1 copy number and higher baseline tryptase.
Non-advanced subtypes (clinical overview)
| Subtype | Typical clinical framing | Why it matters |
|---|---|---|
| Bone marrow mastocytosis (BMM) | Limited marrow involvement; often lower systemic burden | Generally excellent long-term prognosis; still can have mediator symptoms/anaphylaxis risk |
| Indolent systemic mastocytosis (ISM) | No organ damage, but symptoms can be moderate–severe | Quality of life impact can be substantial; targetable KIT biology common |
| Smouldering systemic mastocytosis (SSM) | Higher burden features (without organ damage) | Closer monitoring; may have higher progression risk than ISM |
Targeted care in non-advanced SM: KIT inhibition and the latest evidence
Why target KIT in non-advanced disease?
The targeted therapy: reducing clonal mast cells can lower tryptase and mediator release,
which may translate into improved symptoms and the possibility of disease modification.
- Symptomatic therapy addresses mediator effects (e.g., H1/H2 blockers, leukotriene antagonists, trigger avoidance and emergency plans).
- Targeted KIT inhibition aims to reduce underlying mast cell burden and disease signalling.
Approved targeted option in indolent SM: avapritinib
Randomised evidence from the registrational PIONEER trial demonstrated that avapritinib improved symptom burden versus placebo over 24 weeks,
with objective responses such as ≥50% reductions in serum tryptase in a substantial proportion of patients.
Longer-term follow-up reports continued symptom and quality-of-life improvement with a favourable safety profile in indolent SM.
Regulatory context: the European Commission approved avapritinib for adults with indolent SM with moderate–severe symptoms inadequately controlled on symptomatic treatment in December 2023.
Emerging KIT inhibitors: bezuclastinib (investigational)
Multiple investigational KIT inhibitors are being studied in non-advanced SM. For example, bezuclastinib has reported statistically and clinically meaningful
improvements in symptom scores and reductions in disease measures in clinical trial presentations/abstracts in non-advanced SM.
Note: availability, indications, and commissioning vary by country and over time; always check current local regulatory status and specialist guidance.
and whether targeted therapy is appropriate) should be made by a specialist team familiar with mast cell disorders.
FAQs
How can a patient have non-advanced disease but feel severely unwell?
“Non-advanced” primarily describes absence of organ damage (e.g., liver failure, severe cytopenias from marrow failure).
It does not guarantee low mediator activity. Many patients experience frequent mediator release and high symptom burden affecting daily function.
Is KIT D816V usually inherited?
KIT mutations as predominantly somatic (de novo) in adults, with rare case reports suggesting inherited patterns.
In practice, familial cases are uncommon; specialist genetics input may be considered where there is early onset or a suggestive family history.
Why do some patients do worse even within non-advanced categories?
Risk appears to relate to overall mast cell burden, organ involvement patterns and, in some studies, whether KIT D816V is present across multiple blood cell lineages
(“multilineage involvement”), which may associate with worse long-term outcomes in some cohorts.
References (selected)
- Gotlib J, et al. Avapritinib versus Placebo in Indolent Systemic Mastocytosis. NEJM Evidence (2023).
- Castells M, et al. Continued symptom and quality of life improvement… long-term avapritinib in indolent systemic mastocytosis. JACI: In Practice (2025).
- European Commission approval of avapritinib for indolent SM with moderate–severe symptoms (Dec 2023).
- Jørgensen MP, et al. Prevalence and incidence of mastocytosis in adults (Denmark population study; prevalence as of 2022). (2025).
- British Journal of Haematology: prevalence estimates for mastocytosis in France (2025).
- Orphanet: systemic mastocytosis prevalence estimates in Europe.
- El Hussein S, et al. Systemic Mastocytosis and Other Entities Involving Mast Cells (overview incl. WHO/diagnostic concepts). (2022).
- Bezuclastinib in non-advanced SM: trial/abstract and company presentations (2025).